LJ Institute of Pharmacy - Research Project
December 2022 - April 2023
🧪 What is this Project About?
This research explores how Artificial Intelligence (AI) and Machine Learning (ML) revolutionize chromatographic predictions, particularly in Reverse Phase High-Performance Liquid Chromatography (RP-HPLC). This work focuses on predictive modeling of drug mobile phases, feature engineering, and optimizing chromatographic characteristics.
📌 Why is this Important?
Traditional chromatographic methods require extensive trial-and-error, making the process time-consuming and expensive. With ML models, we can:
- Predict retention times efficiently.
- Optimize chromatographic conditions.
- Reduce experimental costs.
🔗 Research Paper:
Royal Society of Chemistry Publication
🔍 Data Processing & Pipeline
📂 Step 1: Data Collection
The dataset contains chromatographic characteristics of molecules, including:
- Molecular Descriptors (e.g., molecular weight, logP, hydrogen bond acceptors/donors).
- Retention Time Data from various RP-HPLC experiments.
- Chemical Structure Data in SMILES format.
- Molecular Fingerprints generated to analyze structural similarities.
- PubChem & RDKit Extracted Features to enhance predictive accuracy.
🔄 Step 2: Data Cleaning, Feature Engineering & Augmentation
Why is this important?
- Raw datasets often have incomplete or missing molecular properties.
- Standardizing chemical structures ensures consistency in ML models.
- Feature engineering helps extract the most relevant molecular properties.
- Augmenting data increases model robustness and generalization.
🔍 Extracting Relevant Molecular Data
To enhance our dataset, I built a data pipeline to fetch crucial molecular properties from RDKit and PubChem. Each molecule (SMILES format) was processed as follows:
-
Extracting Molecular Descriptors from RDKit
- Molecular weight (MW): Indicates size and bulkiness.
- LogP (lipophilicity): Impacts retention time in chromatography.
- Number of Hydrogen Bond Acceptors/Donors: Influences interaction with the column.
- Topological Polar Surface Area (TPSA): Determines molecular polarity.
from rdkit import Chem from rdkit.Chem import Descriptors def get_rdkit_features(smiles): mol = Chem.MolFromSmiles(smiles) return { "MolecularWeight": Descriptors.MolWt(mol), "LogP": Descriptors.MolLogP(mol), "HBA": Descriptors.NumHAcceptors(mol), "HBD": Descriptors.NumHDonors(mol), "TPSA": Descriptors.TPSA(mol), } data["rdkit_features"] = data["SMILES"].apply(get_rdkit_features) -
Fetching Additional Molecular Properties from PubChem
- Heavy Atom Count: Important for interaction strength.
- Rotatable Bonds: Affects molecular flexibility.
- XLogP: An alternative logP descriptor improving retention prediction.
- Canonical SMILES: Ensures structural standardization.
import requests def get_pubchem_features(smiles): url = f"https://pubchem.ncbi.nlm.nih.gov/rest/pug/compound/smiles/{smiles}/property/MolecularWeight,XLogP,HeavyAtomCount,RotatableBondCount,CanonicalSMILES/JSON" response = requests.get(url).json() props = response["PropertyTable"]["Properties"][0] return { "PubChem_MW": props["MolecularWeight"], "XLogP": props["XLogP"], "HeavyAtomCount": props["HeavyAtomCount"], "RotatableBonds": props["RotatableBondCount"], "CanonicalSMILES": props["CanonicalSMILES"], } data["pubchem_features"] = data["SMILES"].apply(get_pubchem_features) -
Generating Molecular Fingerprints for Structural Similarity Analysis
- Used Morgan Fingerprints to quantify molecular similarity.
- Essential for clustering molecules with similar chromatographic behavior.
from rdkit.Chem import AllChem import numpy as np def get_morgan_fingerprint(smiles, radius=2, nBits=1024): mol = Chem.MolFromSmiles(smiles) fp = AllChem.GetMorganFingerprintAsBitVect(mol, radius, nBits=nBits) return np.array(fp) data["MorganFingerprint"] = data["SMILES"].apply(get_morgan_fingerprint)
🏗️ Feature Engineering & Data Cleaning
Cleaning Techniques Used:
- Handling missing data using mean/mode imputation for continuous variables.
- Standardization & Normalization using Min-Max scaling and Z-score normalization.
- Outlier removal using IQR-based filtering to ensure consistency.
from sklearn.preprocessing import StandardScaler
scaler = StandardScaler()
data_scaled = scaler.fit_transform(data)Feature Selection Using Discriminant Analysis
- Tested multiple combinations of features to determine the most impactful ones.
- Applied Variance Thresholding to remove redundant columns.
- Used Principal Component Analysis (PCA) to reduce dimensionality while retaining critical information.
from sklearn.decomposition import PCA
pca = PCA(n_components=10)
data_pca = pca.fit_transform(data)Data Augmentation Techniques
To expand our dataset and improve generalization, we applied:
- Gaussian Noise Injection to retention time values.
- Molecular Structure Variants generated via SMILES augmentation.
import numpy as np
data['retention_time'] += np.random.normal(0, 0.1, size=len(data))This robust data pipeline significantly improved the accuracy of downstream ML models by providing cleaner and more informative feature sets. The combination of RDKit & PubChem-derived features, molecular fingerprints, and carefully engineered augmentations enabled better chromatographic predictions. 🚀
🔬 Feature Engineering & Selection
🎯 Which Features Mattered Most?
Feature importance was assessed using:
- Correlation Matrix
- SHAP (SHapley Additive exPlanations)
- Recursive Feature Elimination (RFE)
| Feature | Contribution |
|---|---|
| Molecular Weight | ✅ High |
| LogP | ✅ High |
| Hydrogen Bond Acceptors | ❌ Low |
| Ring Count | ✅ Medium |
from sklearn.feature_selection import RFE
selector = RFE(estimator, n_features_to_select=10, step=1)
X_selected = selector.fit_transform(X, y)What Mattered & What Didn’t?
Effective Features:
- Molecular Weight (highly correlated with retention time)
- logP (solubility and interaction with mobile phase)
- Hydrogen Bond Acceptors/Donors (affect polarity)
Ineffective Features:
- Complex ring systems (did not contribute significantly)
- Excessively large molecular descriptors (redundant information)
To filter out irrelevant features, I used Discriminant Analysis:
from sklearn.feature_selection import SelectKBest, f_regression
selector = SelectKBest(score_func=f_regression, k=10)
data_selected = selector.fit_transform(data, target)🔗 Molecular Fingerprints for Structural Analysis
🏷️ Why Use Fingerprints?
- Detecting Structural Similarities
- Grouping Compounds with Similar Retention
We used ECFP4 and MACCS fingerprints to transform molecular structures into numerical representations.
from rdkit import Chem
from rdkit.Chem import AllChem
mol = Chem.MolFromSmiles('CCO')
fingerprint = AllChem.GetMorganFingerprintAsBitVect(mol, 2, nBits=1024)🏗️ Model Development & Training
🛠️ Models Tested
| Model | Performance (R²) |
|---|---|
| Linear Regression | 0.78 |
| Random Forest | 0.85 |
| XGBoost | 0.91 |
| Neural Network | 0.88 |
from xgboost import XGBRegressor
model = XGBRegressor(n_estimators=100, learning_rate=0.05)
model.fit(X_train, y_train)🔗 Overcoming Challenges with Ensemble Learning
Problem: Individual models had limitations in generalizing to new data.
Solution: Implementing stacked ensemble learning combining Random Forest, XGBoost, and ANN.
from sklearn.ensemble import StackingRegressor
ensemble_model = StackingRegressor([
('rf', RandomForestRegressor()),
('xgb', XGBRegressor()),
('nn', MLPRegressor())
])
ensemble_model.fit(X_train, y_train)🎯 Key Takeaways & Future Work
✅ What Worked?
- Feature selection via SHAP & RFE improved model accuracy.
- Data augmentation & molecular fingerprints enhanced predictions.
- Stacked ensembles outperformed individual models.
❌ What Didn’t Work?
- Using all molecular descriptors led to overfitting.
- Linear models failed to capture complex chromatographic relationships.
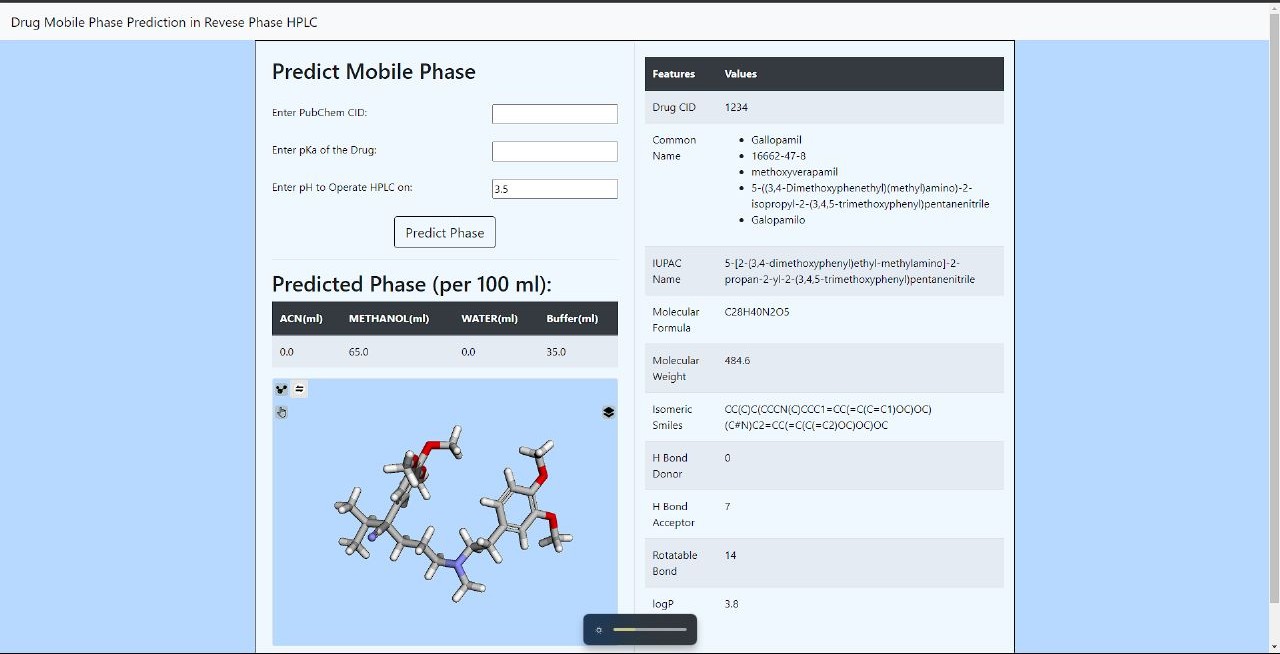
🖥️ Flask-Based UI for Molecular Prediction
I built a Flask web application to allow users to input molecular details and obtain predictions. The UI includes:
- Molecule Description Retrieval via PubChem API
- 3D Molecule Visualizer (RDKit + Py3Dmol integration)
- Predicting Mobile Phase Composition
🔹 How to Use
- Enter the PubChem CID or SMILES string.
- Provide the pKa and pH conditions.
- Click Predict Phase to get suggested solvent composition.
🔹 Expected Output
- Predicted Mobile Phase Proportions (ACN, Methanol, Water, Buffer)
- Molecular Descriptors Display
- Interactive 3D Molecular Structure
from flask import Flask, request, render_template
app = Flask(__name__)
@app.route('/predict', methods=['POST'])
def predict():
cid = request.form['cid']
# Fetch molecular data from PubChem API
# Perform prediction using trained model
return render_template('result.html', prediction=output)🔮 Future Enhancements
- Using Graph Neural Networks (GNNs) for better molecular representation.
- Implementing an Explainable AI in Chromatography framework for transparency.
📜 References & Links
Links : TODO
Tags :
Date : 28th March, Friday, 2025, (Wikilinks: 28th March, March 25, March, 2025. Friday)
Category : Others